
一例II型Waardenburg综合征的基因突变分析
6页
1、 一例II型Waardenburg综合征的基因突变分析 谭英剑 陈志明 莫 然 黄 昕 杨 勇中国医学科学院、北京协和医学院皮肤病医院遗传病中心、江苏省皮肤病与性病学重点实验室,江苏南京,210042Waardenburg综合征(Waardenburg syndrome, WS)是一种具有较强遗传和临床异质性的综合征疾病,以皮肤、毛发等色素异常,虹膜异色和先天性感音神经性耳聋为主要临床特征。1951年,荷兰眼科医师Waardenburg首次报道该病并正式命名为Waardenburg综合征1。目前已发现WS的主要致病基因有MITF、PAX3、SOX10、EDN3、EDNRB和SNAI22。WS可分为4型,I型WS的特征性表现为内眦外移,II型WS与WS1型类似但不伴有内眦外移,3型WS为在WS1型基础上合并肌肉、骨骼系统畸形,4型WS常伴有先天性巨结肠等肠道病变。WS患者主要因先天性耳聋就诊于耳鼻喉科,皮肤科医师对此病的认识较少。本研究对一例WS患儿进行了外显子组测序分析,为明确WS诊断提供了分子遗传学证据,有利于患者及早进行听力检查、遗传咨询、产前诊断等。1 资料与方法1.1 临床资料
2、 先证者,男,10岁。因面部皮疹7年于2022年8月就诊于我院。患儿3岁起出现弥漫分布的雀斑样褐色色素沉着,间杂色素脱失斑点,边界清楚,无隆起,随年龄增长逐渐增多,上肢和臀部也散在色素沉着斑。年幼时头发色素减退,颜色偏黄,后逐渐呈棕黄色。出生时双眼虹膜色素减退,呈天蓝色,后期颜色无明显变化,眼距正常(图1)。出生时听力障碍,2岁行人工耳蜗植入。无生长发育障碍。患儿父母和2个姐姐均正常,父母否认近亲结婚。图1 Waardenburg综合征先证者主要临床表现 1a:面部褐色色素沉着伴色素脱失斑;1b:眼虹膜呈天蓝色;1c:上肢散在色素沉着斑 图2 Waardenburg综合征患者家系及Sanger测序图 2a:家系图;2b:先证者MITF基因7号外显子存在一杂合突变c.649_651delAGA,其父母未检出突变体格检查:一般情况良好,身高146 cm,体重32 kg,体质指数15.0,智力和体格发育无明显异常。听力检查:佩戴人工耳蜗,听力正常。皮肤科检查:面部弥漫分布黄豆大褐色色素沉着斑,间杂色素脱失斑点,上肢和臀部散在分布类似褐色斑点。头发色素减退,呈棕黄色。眼科检查:双眼虹膜色素减退
3、,呈天蓝色,眼距和视力正常。1.2 方法1.2.1 遗传性皮肤病目标基因外显子组测序 本研究经中国医学科学院皮肤病医院医学伦理委员会批准(2019)临审第(005)号,选取先证者及其父母作为基因突变检测对象,三人均签署知情同意书后,抽取外周血各2 mL置于真空EDTA抗凝管内,提取基因组DNA,送至北京迈基诺基因科技股份有限公司行遗传性皮肤病目标基因外显子组测序,目标区域长度为2.46 Mb,含已知致病基因726个。数据下机后,从该患者DNA中总共检测出5188个SNP变异位点,对其进行分析筛选:第一步筛选位于外显子区或剪切区的位点,得到1545个位点;第二步选择最高人群频率0.05的位点,得到127个位点;第三步过滤掉质控数据较差的突变,剩余101个位点;第四步筛除Clinvar数据库中已知的良性位点,剩下可能致病的或不确定的位点,同时选择Disease Information中存在对应疾病的位点,剩余38个位点;第五步筛选突变频率0.2,测序深度20的位点,得到32个位点;最后选择其中符合临床表型的致病基因,进行下一步分析。1.2.2 Sanger测序 根据检测得到的可疑致病突变位
《一例II型Waardenburg综合征的基因突变分析》由会员金***分享,可在线阅读,更多相关《一例II型Waardenburg综合征的基因突变分析》请在金锄头文库上搜索。

一场饱含行业情怀的纤维革新之旅

一例黄体期长方案降调节中多卵泡发育的病例报道及文献分析

一元一次不等式与一次函数课例探析

一例恶性黑色素瘤患者使用卡度尼利单抗导致的免疫性心肌炎的个案护理-第1篇
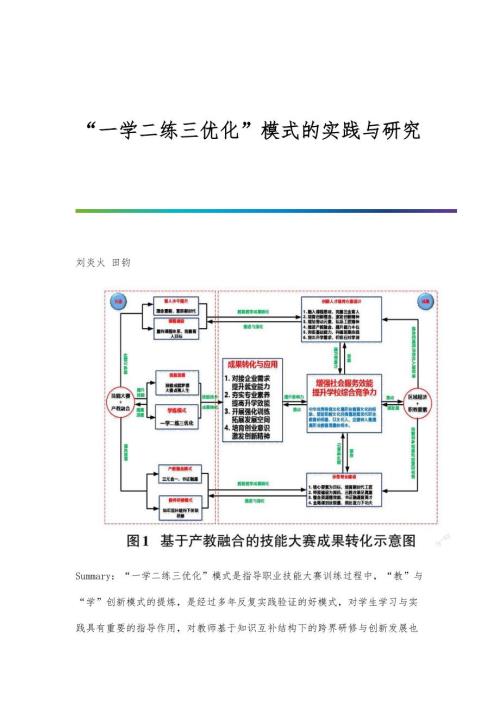
一学二练三优化模式的实践与研究

一医一药一患药学服务模式的效果评价研究

一对一带教双轨教学在产科学实习中的应用价值分析

一例三阴性终末期乳腺癌患者的安宁疗护实践

一对一责任制助产护理对减少产妇不良情绪及促进妊娠的效果分析

一堂新授课的教学设计探究-直线与圆的位置关系

一元二次方程教学实践中的逆向思维研究

一小的整体解决离不开高质量的儿童友好产业

一例典型道路交通事故车辆灯光使用情况鉴定案例分析

一体式主动脉弓分支重建支架治疗累及左锁骨下动脉的主动脉穿透性溃疡疗效分析△

一例II型Waardenburg综合征的基因突变分析

一家企业集团如何通过司库体系打好金融牌
一带一路倡议下东北亚经济合作研究

一场美读和项目式学习的双向奔赴-以《阿房宫赋》教学为例

一例犬髋关节脱位的诊断治疗

一体式蔬菜钵苗取苗爪夹持力检测传感器设计与试验
 虚拟现实技术在仪表培训中的创新
虚拟现实技术在仪表培训中的创新
2024-05-22 22页
虚拟现实增强现实在质量管理中的应用
2024-05-22 27页
虚拟现实和增强现实技术在学术图书馆中的应用
2024-05-22 26页
虚拟现实技术在仪表故障树分析中的应用
2024-05-22 22页
虚拟现实体验设计最佳实践
2024-05-22 25页
虚拟现实技术在仪表教育中的应用
2024-05-22 23页
虚拟现实和增强现实技术在物流培训中的应用
2024-05-22 24页
虚拟现实和增强现实技术在建筑营销中的应用
2024-05-22 23页
虚拟现实与增强现实技术在美术中的应用
2024-05-22 23页
虚拟现实和增强现实技术在物业管理中的应用
2024-05-22 23页