
ISO 13485 2016与GMP差异对比
30页
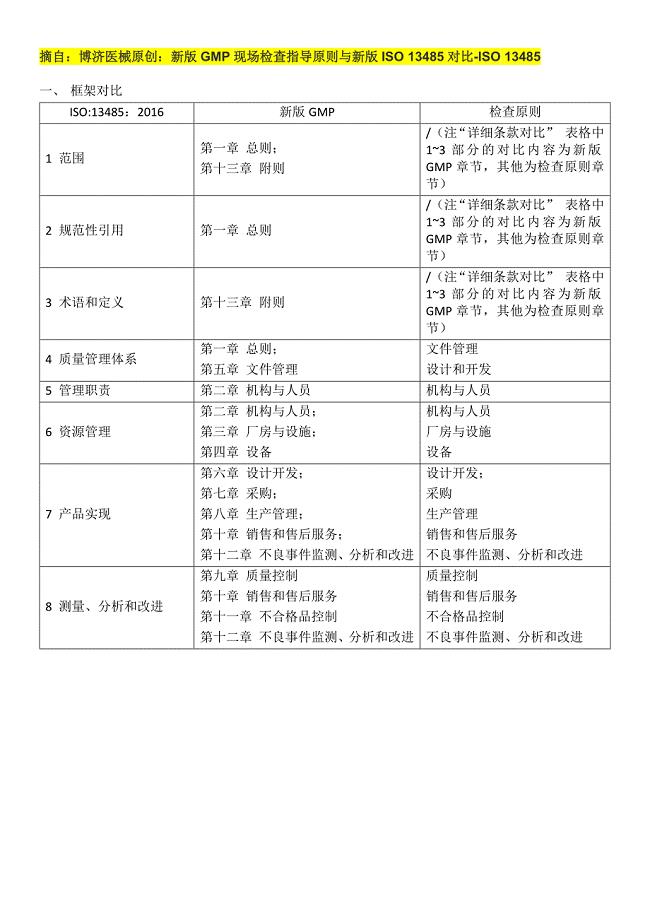
1、摘自:博济医械原创:新版GMP现场检查指导原则与新版ISO 13485对比-ISO 13485一、 框架对比 ISO:13485:2016新版GMP检查原则1 范围第一章 总则;第十三章 附则/(注“详细条款对比” 表格中13部分的对比内容为新版GMP章节,其他为检查原则章节)2 规范性引用第一章 总则/(注“详细条款对比” 表格中13部分的对比内容为新版GMP章节,其他为检查原则章节)3 术语和定义第十三章 附则/(注“详细条款对比” 表格中13部分的对比内容为新版GMP章节,其他为检查原则章节)4 质量管理体系第一章 总则;第五章 文件管理文件管理设计和开发5 管理职责第二章 机构与人员机构与人员6 资源管理第二章 机构与人员;第三章 厂房与设施;第四章 设备机构与人员厂房与设施设备7 产品实现第六章 设计开发;第七章 采购;第八章 生产管理;第十章 销售和售后服务;第十二章 不良事件监测、分析和改进设计开发;采购生产管理销售和售后服务不良事件监测、分析和改进8 测量、分析和改进第九章 质量控制第十章 销售和售后服务第十一章 不合格品控制第十二章 不良事件监测、分析和改进质量控制销
2、售和售后服务不合格品控制不良事件监测、分析和改进二、 详细条款对比 ISO 13485:2016新版GMP/检查原则差异浅析1范围本标准明确了质量管理体系的要求,组织需要说明能够提供一直符合客户和适用法规要求的医疗器械和相关服务。医疗器械生产质量管理规范第六条企业负责人是医疗器械产品质量的主要责任人,应当履行以下职责;(四)按照法律、法规和规章的要求组织生产。第三十一条 设计和开发输出应当满足输入要求,包括采购、生产和服务所需的相关信息、产品技术要求等。设计和开发输出应当得到批准,保持相关记录;第七条 企业负责人应当确定一名管理者代表。管理者代表应当建立、实施并保持质量管理体系,报告质量管理体系的运行情况和改进需求,提高员工满足法规、规章和顾客要求的意识。无重大差异这些组织可为产品生命周期中的一个或多个阶段的组织,包括医疗器械的设计开发、生产、储存和销售、安装或服务,以及相关活动的设计开发和提供(如技术支持)。这一国际标准也可用于提供产品的供应商或外来方,包括向组织提供质量管理体系相关服务。除非明确声明,本国际标准的要求适用于所有大小和所有类型的企业,明确适用于医疗器械的要求,也等同适
3、用于组织提供的相关服务第二条 医疗器械生产企业(以下简称企业)在医疗器械设计开发、生产、销售和售后服务等过程中应当遵守本规范的要求。第七十九条 医疗器械注册申请人或备案人在进行产品研制时,也应当遵守本规范的相关要求。1. 新版GMP的适用对象未明确为提供产品的供应商或外来方,但在“第七章 采购”,强调了企业应审核供应商(包括产品和服务),企业可参考医疗器械生产企业供应商审核指南;2. 13485明确强调,即企业可要求供应商或外来方进行13485验证,或者根据13485晚上供应商或外来方的体系;3. 13485的适用范围更广,而GMP范围较窄,较适用于医疗器械生产企业,医疗器械经营企业需要参考医疗器械经营质量管理规范4. 其余差异为各国的注册/认证流程和形式的差异所致,无重大区别。其余内容简述:详述了企业如何处理不适用的条款,并提供相关说明。第八十一条 企业可根据所生产医疗器械的特点,确定不适用本规范的条款,并说明不适用的合理性。无2规范性引用ISO 9000:2015医疗器械监督管理条例(国务院第650号);医疗器械生产监督管理办法(国家食品药品监督管理总局令第7号)因各国法规监管体系
4、而异3 术语和定义忠告性通知、授权代表、临床评价、抱怨、销售、植入医疗器械、进口商、标示资料、生命周期、生产者、医疗器械、医疗器械族、性能评价、上市后监督、产品、采购产品、风险、风险管理、无菌屏障系统、无菌医疗器械第八十二条验证、确认、关键工序、特殊过程1. 13485虽未提及GMP的定义,但ISO 9000:2015均有详细定义;2. 在适用法规要求的前提下,GMP中术语可参考YY/T 0287-2003及其他相关国家或行业标准。4质量管理体系4.1一般要求4.1.1组织应按本标准要求和适用法规的要求,建立质量管理体系,形成文件并维持其有效性;组织应建立、实施和维持本标准或适用法规要求形成文件的要求,程序、活动或安排;组织应对符合适用法规要求的组织担任角色形成文件。注:组织担任角色包括生产企业、授权代表、进口商或销售企业。第三条 企业应当按照本规范的要求,结合产品特点,建立健全与所生产医疗器械想适应的质量管理体系,并保证其有效运行;第六条 企业负责人是医疗器械产品质量的主要责任人,应当履行以下职责:(四)按照法律、法规和规章的要求组织生产。13485的适用对象更广。4.1.2组织应当
《ISO 13485 2016与GMP差异对比》由会员伟、分享,可在线阅读,更多相关《ISO 13485 2016与GMP差异对比》请在金锄头文库上搜索。














 心内一科降低住院患者静脉输液使用率实施方案
心内一科降低住院患者静脉输液使用率实施方案
2024-04-17 4页
院降低住院患者静脉输液率PDCA
2024-04-17 18页
急诊医学分娩及其并发症病案分析病例讨论
2024-02-25 8页
急诊医学电复律和临时起搏病案分析病例讨论
2024-02-25 4页
急诊医学单纯疱疹病案分析病例讨论
2024-02-25 2页
急诊医学基本穿刺技术病案分析病例讨论
2024-02-25 5页
急诊医学肠系膜缺血病案分析病例讨论
2024-02-25 2页
急诊医学多形红斑病案分析病例讨论
2024-02-25 2页
急诊医学抽搐病案分析病例讨论
2024-02-25 7页
急诊医学急性冠脉综合征病案分析病例讨论
2024-02-25 4页