
医疗器械监督管理条例
14页
1、医疗器械监督管理条例(2000年1月4日中华人民共和国国务院令第276号公布2014年2月12日国务院第39次常务会议修订通过根据2017年5月4日国务院关于修改医疗器械监督管理条例的决定修订)第一章总则第一条为了保证医疗器械的安全、有效,保障人体健康和生命安全,制定本条例。第二条在中华人民共和国境内从事医疗器械的研制、生产、经营、使用活动及其监督管理,应当遵守本条例。第三条国务院食品药品监督管理部门负责全国医疗器械监督管理工作。国务院有关部门在各自的职责范围内负责与医疗器械有关的监督管理工作。县级以上地方人民政府食品药品监督管理部门负责本行政区域的医疗器械监督管理工作。县级以上地方人民政府有关部门在各自的职责范围内负责与医疗器械有关的监督管理工作。国务院食品药品监督管理部门应当配合国务院有关部门,贯彻实施国家医疗器械产业规划和政策。第四条国家对医疗器械按照风险程度实行分类管理。第一类是风险程度低,实行常规管理可以保证其安全、有效的医疗器械。第二类是具有中度风险,需要严格控制管理以保证其安全、有效的医疗器械。第三类是具有较高风险,需要采取特别措施严格控制管理以保证其安全、有效的医疗器械
2、。 评价医疗器械风险程度,应当考虑医疗器械的预期目的、结构特征、使用方法等因素。国务院食品药品监督管理部门负责制定医疗器械的分类规则和分类目录,并根据医疗器械生产、经营、使用情况,及时对医疗器械的风险变化进行分析、评价,对分类目录进行调整。制定、调整分类目录,应当充分听取医疗器械生产经营企业以及使用单位、行业组织的意见,并参考国际医疗器械分类实践。医疗器械分类目录应当向社会公布。第五条医疗器械的研制应当遵循安全、有效和节约的原则。国家鼓励医疗器械的研究与创新,发挥市场机制的作用,促进医疗器械新技术的推广和应用,推动医疗器械产业的发展。第六条医疗器械产品应当符合医疗器械强制性国家标准;尚无强制性国家标准的,应当符合医疗器械强制性行业标准。一次性使用的医疗器械目录由国务院食品药品监督管理部门会同国务院卫生计生主管部门制定、调整并公布。重复使用可以保证安全、有效的医疗器械,不列入一次性使用的医疗器械目录。对因设计、生产工艺、消毒灭菌技术等改进后重复使用可以保证安全、有效的医疗器械,应当调整出一次性使用的医疗器械目录。第七条医疗器械行业组织应当加强行业自律,推进诚信体系建设,督促企业依法开展生
3、产经营活动,引导企业诚实守信。第二章 医疗器械产品注册与备案第八条第一类医疗器械实行产品备案管理,第二类、第三类医疗器械实行产品注册管理。第九条第一类医疗器械产品备案和申请第二类、第三类医疗器械产品注册,应当提交下列资料:(一)产品风险分析资料;(二)产品技术要求;(三)产品检验报告;(四)临床评价资料;(五)产品说明书及标签样稿;(六)与产品研制、生产有关的质量管理体系文件;(七)证明产品安全、有效所需的其他资料。医疗器械注册申请人、备案人应当对所提交资料的真实性负责。第十条第一类医疗器械产品备案,由备案人向所在地设区的市级人民政府食品药品监督管理部门提交备案资料。其中,产品检验报告可以是备案人的自检报告;临床评价资料不包括临床试验报告,可以是通过文献、同类产品临床使用获得的数据证明该医疗器械安全、有效的资料。向我国境内出口第一类医疗器械的境外生产企业,由其在我国境内设立的代表机构或者指定我国境内的企业法人作为代理人,向国务院食品药品监督管理部门提交备案资料和备案人所在国(地区)主管部门准许该医疗器械上市销售的证明文件。备案资料载明的事项发生变化的,应当向原备案部门变更备案。第十一条
4、申请第二类医疗器械产品注册,注册申请人应当向所在地省、自治区、直辖市人民政府食品药品监督管理部门提交注册申请资料。申请第三类医疗器械产品注册,注册申请人应当向国务院食品药品监督管理部门提交注册申请资料。向我国境内出口第二类、第三类医疗器械的境外生产企业,应当由其在我国境内设立的代表机构或者指定我国境内的企业法人作为代理人,向国务院食品药品监督管理部门提交注册申请资料和注册申请人所在国(地区)主管部门准许该医疗器械上市销售的证明文件。第二类、第三类医疗器械产品注册申请资料中的产品检验报告应当是医疗器械检验机构出具的检验报告;临床评价资料应当包括临床试验报告,但依照本条例第十七条的规定免于进行临床试验的医疗器械除外。第十二条受理注册申请的食品药品监督管理部门应当自受理之日起3个工作日内将注册申请资料转交技术审评机构。技术审评机构应当在完成技术审评后向食品药品监督管理部门提交审评意见。第十三条受理注册申请的食品药品监督管理部门应当自收到审评意见之日起20个工作日内作出决定。对符合安全、有效要求的,准予注册并发给医疗器械注册证;对不符合要求的,不予注册并书面说明理由。国务院食品药品监督管理部门
《医疗器械监督管理条例》由会员cn****1分享,可在线阅读,更多相关《医疗器械监督管理条例》请在金锄头文库上搜索。

2023年节能环保管理规定

八年级数学下学期期末复习《勾股定理》课案(学生用) 新人教版

大年初三祝福语集锦
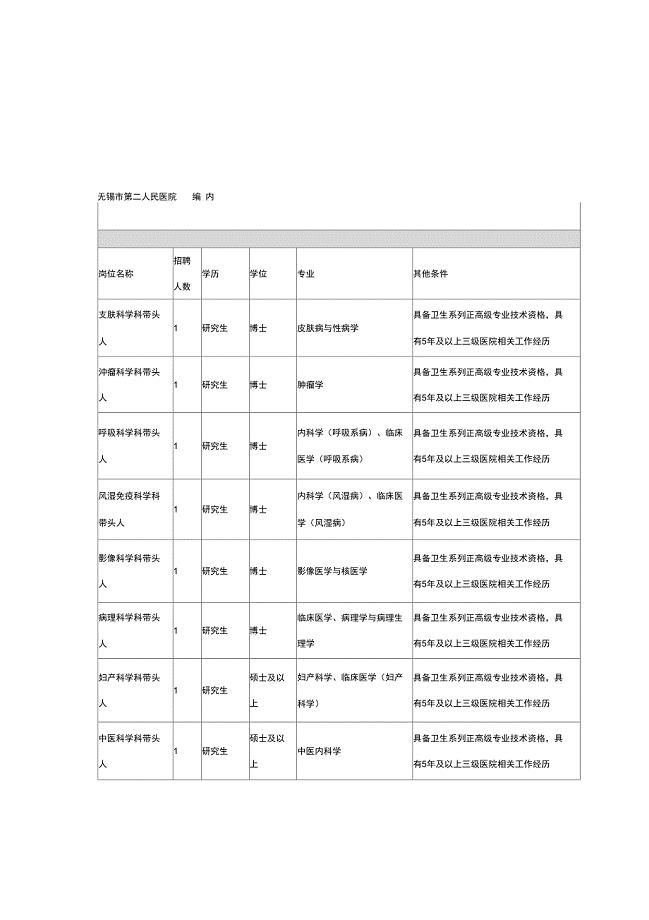
无锡市第二人民医院——编内

2022年考博英语-通用考博英语考试题库及全真模拟冲刺卷(含答案带详解)套卷29

房地产销售个人工作计划标准范文(6篇).doc

人教版五年级下册英语教案(最新)

《圆的面积》教学设计方案

营销与策划求职信汇总七篇

直流电机调速控制系统的C语言程序 (2)

2011届高考生物第一轮复习满分练兵场 5-3 生态系统的类型和结构

初二德育2023个人学期工作计划范本(3篇).doc

培养优生工作计划

质量保证体系及措施

东北大学21秋《常用电器控制技术含PLC》平时作业一参考答案57

2022年我的长征观后感15篇

2019年高考语文冲刺三轮提分练 板块组合滚动练7 语言文字应用+名句名篇默写+论述类文本阅读(含解析)
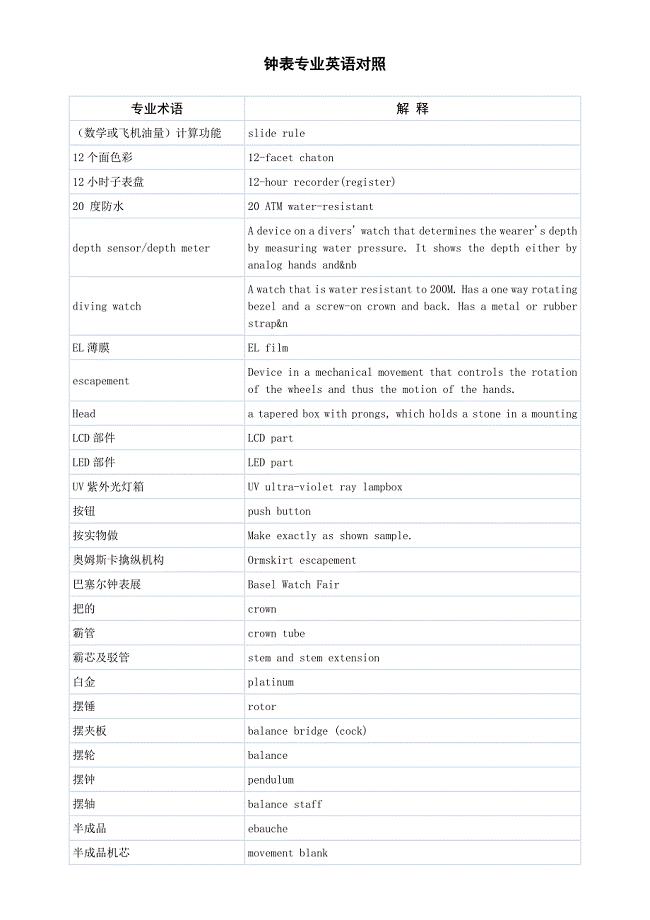
钟表专业英语对照

积的乘方说课稿

个人教育自传
 咸阳光学元器件项目招商引资方案_参考模板
咸阳光学元器件项目招商引资方案_参考模板
2023-02-20 149页
弥渡县公共服务设备项目商业计划书【范文参考】
2024-01-22 124页
幼儿园春节活动方案2022
2023-06-18 14页
土建造价工程师岗位的工作职责(10篇)
2023-06-23 8页
2021年创先争优活动心得体会-关于创先争优的心得体会
2022-10-03 2页
公司和大学新闻传播学院合作合同
2023-04-04 24页
十堰智能驾驶设备项目商业计划书_模板
2023-08-17 134页
内蒙古火电技术服务项目实施方案
2022-10-28 227页
干线光缆割接施工组织方案-传输管线工程
2023-10-30 12页
锡林郭勒盟绿色低碳消费项目招商计划书(参考模板)
2023-05-26 160页