
药企2010新版GMP实用培训材料整理
10页
1、药企2010新版GMP实用培训材料整理主要内容第一部分: 新版GMP简述第二部分: 新版GMP学习体会探讨第三部分: 实施新版GMP的困难第四部分: 新版GMP中术语的含义第一部分:新版GMP简述什么是GMP?* 药品生产质量管理规范* GMP是Good Manufacturing Practice for Drugs的简称。* 是在生产全过程中,用科学、合理、规范化的条件和方法来保证生产优良药品的一整套科学管理方法。* 十四章分别为:第一章总则 第二章质量管理 第三章机构与人员 第四章厂房与设施 第五章设备第六章物料与产品第七章确认与验证 第八章文件管理第九章生产管理第十章质量控制与质量保证 第十一章委托生产与委托检验 第十二章产品发运与召回第十三章自检第十四章附则 建立质量管理体系质量管理 质量保证:是质量管理体系的一部分。企业必须建立质量保证系统,同时建立完整的文件体系,以保证系统有效运行。 质量控制:包括相应的组织机构、文件系统以及取样、检验等,确保物料或产品在放行前完成必要的检验,确认其质量符合要求。 质量风险管理:质量风险管理是在整个产品生命周期中采用前瞻或回顾的方式,对质
2、量风险进行评估、控制、沟通、审核的系统过程。 所采用的方法、措施、形式及形成的文件应当与存在风险的级别相适应。 机构与人员 * 企业应当建立与药品生产相适应的管理机构,并有组织机构图。* 关键人员:包括企业负责人、生产管理负责人、质量管理负责人和质量受权人。 * 企业负责人 :是药品质量的主要责任人,全面负责企业日常管理。 * 生产管理负责人 资质:药学或相关专业本科学历(或中级专业技术职称或执业药师资格),具有至少三年从事药品生产和质量管理的实践经验,其中至少有一年的药品生产管理经验,接受过与所生产产品相关的专业知识培训。 * 质量管理负责人 资质:具有药学或相关专业本科学历(或中级专业技术职称或执业药师资格),具有至少五年从事药品生产和质量管理的实践经验,其中至少一年的药品质量管理经验,接受过与所生产产品相关的专业知识培训。 * 质量受权人 资质:具有药学或相关专业本科学历(或中级专业技术职称或执业药师资格),具有至少五年从事药品生产和质量管理的实践经验,从事过药品生产过程控制和质量检验工作。 机构与人员* 个人卫生的要求* 从药人员应当随时注意个人清洁卫生,进出洁净区严格执行人员
3、进出车间净化、更衣程序;参观人员和未经培训的人员不得进入生产区和质量控制区,特殊情况确须进入的,应当事先对个人卫生、更衣等事项进行指导;任何进入生产区的人员均应当按规定更衣。* 操作人员应当避免裸手接触药品、与药品直接接触的包装材料和设备表面,不可避免时,手部应当及时消毒;生产区、仓储区应当禁止吸烟和饮食,禁止存放食品、饮料、香烟和个人用药品等非生产用物品。厂房与设施 * 厂房的选址、设计、布局、建造、改造和维护必须符合药品生产要求,应当能够最大限度地避免污染、交叉污染、混淆和差错,便于清洁、操作和维护。 * 为降低污染和交叉污染的风险,厂房、生产设施和设备应当根据所生产药品的特性、工艺流程及相应洁净度级别要求合理设计、布局和使用 。* 质量控制实验室通常应当与生产区分开。生物检定、微生物实验室还应当彼此分开。 设备 * 设备的设计、选型、安装、改造和维护必须符合预定用途,应当尽可能降低产生污染、交叉污染、混淆和差错的风险,便于操作、清洁、维护,以及必要时进行的消毒或灭菌。 * 生产设备清洁的操作规程应当规定具体而完整的清洁方法、清洁用设备或工具、清洁剂的名称和配制方法、去除前一批次标
4、识的方法、保护已清洁设备在使用前免受污染的方法、已清洁设备最长的保存时限、使用前检查设备清洁状况的方法,使操作者能以可重现的、有效的方式对各类设备进行清洁。* 应当按照操作规程和校准计划定期对生产和检验用衡器、量具、仪表、记录和控制设备以及仪器进行校准和检查,并保存相关记录。 * 制药用水 :饮用水、纯化水、注射用水。应当按照操作规程对纯化水、注射用水管道进行清洗消毒,并有相关记录。发现制药用水微生物污染达到警戒限度、纠偏限度时应当按照操作规程处理。 物料与产品 * 应当建立物料和产品的操作规程,确保物料和产品的正确接收、贮存、发放、使用和发运,防止污染、交叉污染、混淆和差错。* 所有到货物料均应当检查,以确保与订单一致,并确认供应商已经质量管理部门批准。 * 不合格的物料、中间产品、待包装产品和成品的每个包装容器上均应当有清晰醒目的标志,并在隔离区内妥善保存。处理应当经质量管理负责人批准,并有记录。 确认与验证 * 企业应当制定验证总计划,以文件形式说明确认与验证工作的关键信息。 * 确认或验证应当按照预先确定和批准的方案实施,并有记录。确认或验证工作完成后,应当写出报告,并经审核、
《药企2010新版GMP实用培训材料整理》由会员jiups****uk12分享,可在线阅读,更多相关《药企2010新版GMP实用培训材料整理》请在金锄头文库上搜索。

建立安全生产长效机制-共创和谐平安输气管道

土石方爆破工程设计方案

康师傅百货商场制度汇编之退换货管理办法

庙头中学2012年中考百日誓师大会学生代表发言稿

围堰拆除爆破工程

华能洱源马鞍山风电场工程土石方爆破管理制度

国资委:全面开展管理提升活动-为培育世界一流企业奠定坚实基础

《专业技术人员职业发展与规划》电子书

应收票据审计方案
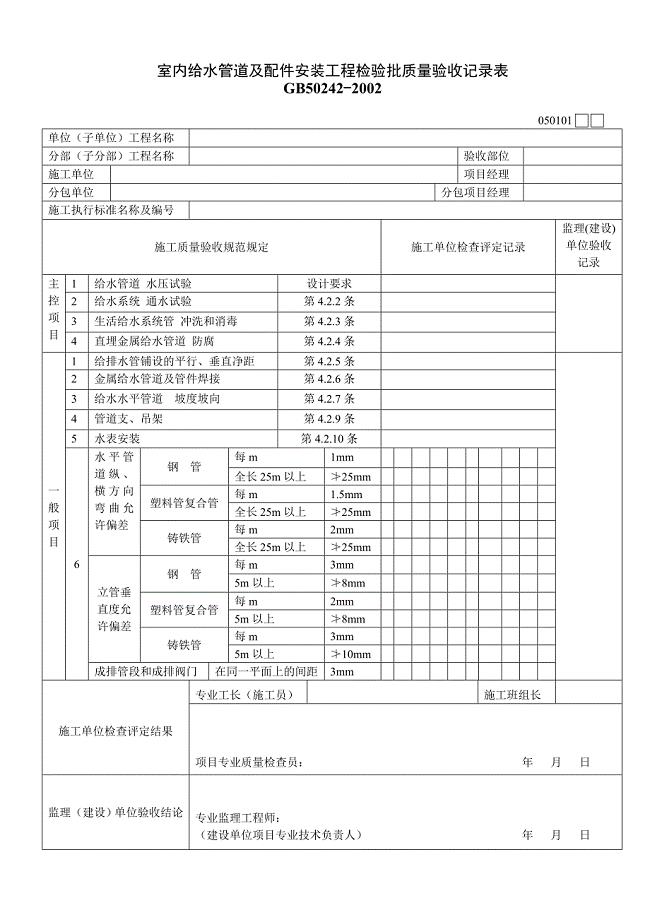
建筑给水排水与采暖工程质量验收用表

广播电视管理条例行政处罚裁量标准

广东电力系统调度运行操作管理规定

建筑给排水工程名词解释

幼儿园小班安全健康活动教案:宝宝误食后的急救

建业集团房屋建筑工程交房标准内容技术交底

建筑心理学论文(1)

康师傅百货商场制度汇编之工服管理程序009

建筑工程专业一级建造师继续教育培训结业报告20

廉洁风险防控回头看工作汇报材料

平台工作人员服务规范
 2024年安徽省芜湖市中考二模语文试卷【含答案】
2024年安徽省芜湖市中考二模语文试卷【含答案】
2024-04-24 10页
2024届河北省邯郸市中考一模语文试题【含答案】
2024-04-24 10页
2024年(6月份)中考数学押题试卷【含答案】
2024-04-24 25页
2024年江苏省扬州市宝应县中考一模语文试题【含答案】
2024-04-24 10页
辽宁省本溪市2022-2023学年高中下学期学业水平考试美术试题【含答案】
2024-04-23 4页
第六单元 正比例和反比例 (单元测试卷)苏教版数学六年级下册【含答案】
2024-04-23 10页
江苏省无锡市2024年七年级下学期期中数学调研试卷【含答案】
2024-04-23 19页
江苏省江阴市华士片2022-2023学年七年级下学期期中语文试题【含答案】
2024-04-23 10页
江苏省泰州兴化市2023-2024学年高一下学期期中考试语文试题【含答案】
2024-04-23 16页
小学六年级体育与健康测试题【含答案】
2024-04-23 5页
审定新标准一年级起点新外研社版小学三年级下册英语全册mp3音频课文朗读下载薪酬培训演说稿二年级下册英语试题期中考查--牛津上海版2021年抢劫案件开庭审理笔录新编2023年浙江温州中考数学试题及答案上大继续教育专升本中级管理学-平时作业某央企干部国有经济和国有企业高质量发展研学班个人总结某市税务局扎实开展“五星”机关党组织创建推动县级税务局政治机关建设上台阶见实效经验总结材料某县税务局加强县级政治机关建设推进党业融合工作总结汇报材料某央企干部2024年国有经济和国有企业高质量发展研学班个人总结某区委书记在区高质量发展总结表彰大会上的讲话某镇党委落实党风廉政建设责任制自查自评报告