
遗传学:生化遗传学
73页
1、生化遗传学,基因决定蛋白质的合成,基因突变,基因突变导致蛋白质合成异常引起的疾病,蛋白缺陷,机体病变,分 子 病,基因决定蛋白质的合成,血红蛋白病,血浆蛋白病,酶蛋白病,受体蛋白病,膜转运载体蛋白病,10.1 血红蛋白及其基因突变导致的疾病,发病率较高,地 位 特 殊,估计全世界血红蛋白致病基因携带者超过5%,蛋白结构最早被揭示,人类珠蛋白基因最早被克隆,分子和生化变异机制被清楚揭示。,10.1.1 正常人体血红蛋白的结构和遗传控制,6种: 、(G、A)、,血红蛋白功能,血红蛋白结构,珠蛋白肽链类型,机体输送氧气,肽链+血红素 血红蛋白单体4 血红蛋白,10.1.1 正常人体血红蛋白的结构和遗传控制,高度保守 链42位苯丙氨酸和链92位组氨酸,珠蛋白肽链,珠蛋白肽链,和铁相结合的位置,链长141个氨基酸,、类链长146个氨基酸,10.1.1 正常人体血红蛋白的结构和遗传控制,血红蛋白类型,10.1.1 正常人体血红蛋白的结构和遗传控制,珠蛋白基因簇,珠蛋白基因簇,珠蛋白基因结构, 基因, 基因, 基因, 基因,基因,基因,10.1.1 正常人体血红蛋白的结构和遗传控制,珠蛋白基因结构,
2、10.1.1 正常人体血红蛋白的结构和遗传控制,珠蛋白基因簇,珠蛋白基因簇,10.1.1 正常人体血红蛋白的结构和遗传控制,位点控制区 (-LCR): 16号染色体短臂基因5端上游40kb处。,位点控制区 (-LCR):11号染色体短臂基因上游20kb处。,LCR作用:调控基因正常表达量和表达时相,10.1.1 正常人体血红蛋白的结构和遗传控制,提供一个开放的染色质区域,使转录因子与基因簇中调节元件接触; 它是基因簇中基因转录的超级增强子,LCR调控机制:,10.1.1 正常人体血红蛋白的结构和遗传控制,和珠蛋白基因簇中53基因的排列顺序与它们在个体发育中的表达顺序相同。,10.1.2 血红蛋白结构变异型的遗传效应,引起溶血性贫血的血红蛋白结构变异型 影响氧转运的血红蛋白结构变异型 造成地中海贫血样表现的血红蛋白结构变异型,血红蛋白结构变异型,血 红 蛋 白 病,血红蛋白结构变异型类型,血红蛋白基因突变后改变珠蛋白的结构,1000多个血红蛋白结构变异型,近半数可致病,10.1.2.1 引起溶血性贫血的血红蛋白结构变异型,HbS即镰状细胞血红蛋白,镰状红细胞变形能力下降,单个细胞不能通过
3、毛细血管,血流被阻断,局部缺血。,溶解度只有正常的1/5,在低氧分压状态下HbS分子聚集成杆状多聚体或纤维状,使红细胞镰变,AR遗传病,杂合子个体兼有两种血红蛋白HbA ( 2A 2)和HbS( 2S 2),主要分布在非洲,散发于地中海和印度,链第6位谷氨酸被缬氨酸取代(GAGGTG: 谷缬),10.1.2.1 引起溶血性贫血的血红蛋白结构变异型,溶解度降低,结晶析出,降低了细胞的变形功能,不能通过毛细血管,引起中等程度溶血。,AR遗传疾病,纯合子罹患HbC病。,C的频率在西非很高,链第6位密码子单个碱基突变造成,使 谷氨酸被赖氨酸取代(谷赖),HbC,10.1.2.1 引起溶血性贫血的血红蛋白结构变异型,四聚体不可溶解,沉淀形成包涵体,即Heinz小体(Heinz body),破坏红细胞膜引起溶血,取代后,血红素易脱离,造成不稳定,Hb Hammersmith 的氧亲和力也降低,会引起青紫(cyanosis)。,Hb Hammersmith,链第42位苯丙氨酸被丝氨酸取代,突变后链可形成空间结构,但血红蛋白很不稳定,容易导致溶血。,10.1.2.1 引起溶血性贫血的血红蛋白结构变异型
4、,基因有15 bp的缺失,珠蛋白基因开放读码框不变,但缺失了9195位5个氨基酸残基。,Hb Gun Hill,10.1.2.2 影响氧转运的血红蛋白结构变异型,不能携氧的 高铁血红蛋白 (methemoglobin),(oxygen affinity),增高或降低血红蛋白的氧亲和力,10.1.2.2 影响氧转运的血红蛋白结构变异型,Hb Hyde park,Hb Kempsey和Hb Kansas,92位高度保守的组氨酸被酪氨酸取代,产生高铁血红蛋白。,Hb Kempsey(99天冬氨酸天冬酰胺),血红蛋白处疏松状态,氧亲和力很高,导致红细胞增多症。 Hb Kansas (99天冬氨酸苏氨酸),血红蛋白不能变为疏松状态,氧亲和力很低。携带者有青紫。,10.1.2.2 影响氧转运的血红蛋白结构变异型,链146和147位密码子(CACUAA)之间插入 2个碱基AC,导致移码突变,147位终止密码UAA 变成苏氨酸的密码ACU,链的长度延长到157个 氨基酸,肽链氧亲和力随之增高。,Hb Tak,10.1.2.3造成地中海贫血表现的血红蛋白结构变异型,HbE,大多数此类突变主要影响 mRN
《遗传学:生化遗传学》由会员hu****a8分享,可在线阅读,更多相关《遗传学:生化遗传学》请在金锄头文库上搜索。

《小学生防溺水安全教育班会》教案

《中小学预防传染病知识主题班会》教案

《幼儿园防溺水安全教育班会》教案模板

小学开学第一课主题班会教案八篇

垃圾分类主题班会《保护环境 美化校园》教案

小学开学第一课主题班会教案和含演讲稿
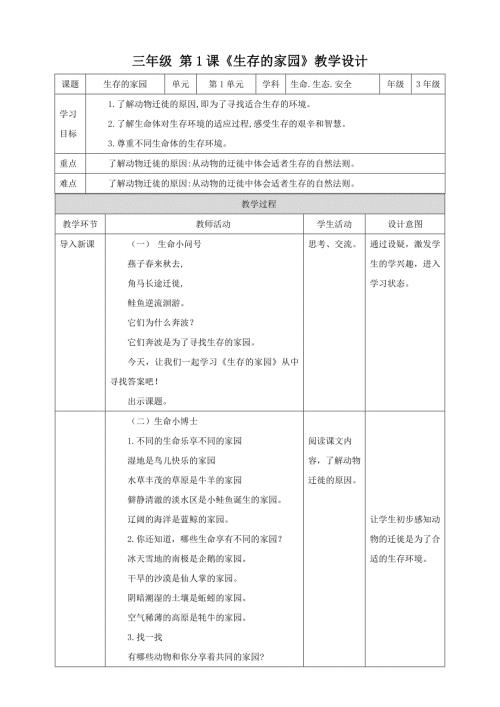
第1课《生存的家园》教学设计
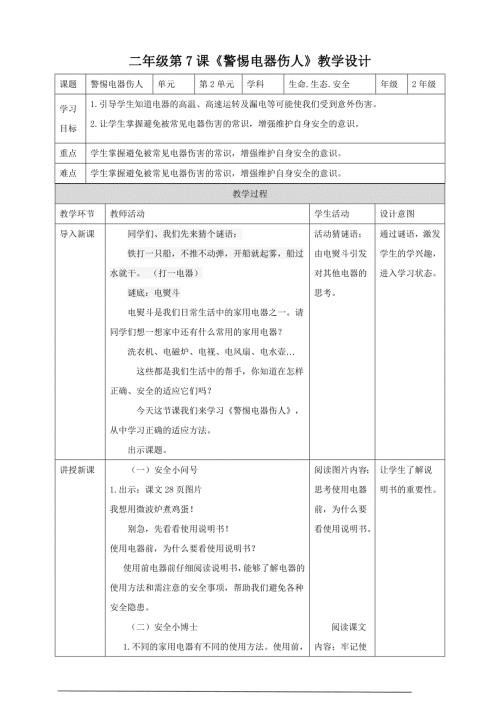
第7课《警惕电器伤人》教学设计
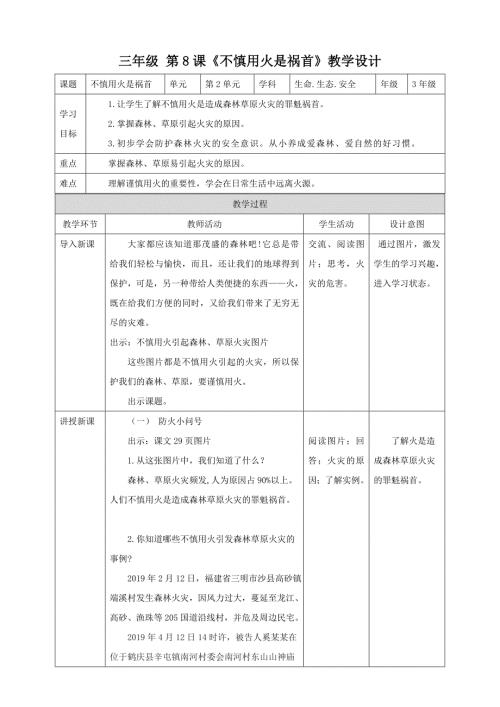
第8课《不慎用火是祸首》教学设计
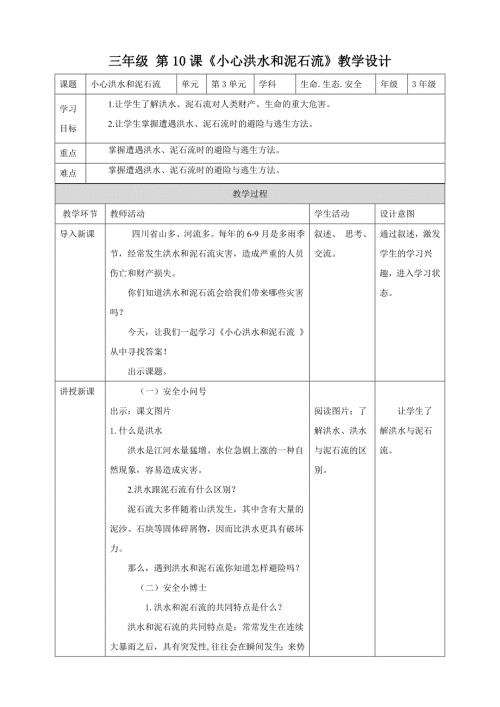
第10课《小心洪水和泥石流》教学设计
第11课《及时治小伤》教学设计
第1课《身体里的“乐队”》教案
第6课《校园避震有办法》教学设计
第18课《有你有我真温暖》教学设计
第13课《每天锻炼一小时》教学设计
第14课《我与动物亲密有间》教学设计
第16课《我行我能行》教学设计
第12课《儿童肥胖的危害》教学设计

第17课《美丽的插花》教学设计

第8课《我和规则做朋友》教学设计
 突发新冠疫情封闭式管理应急预案培训
突发新冠疫情封闭式管理应急预案培训
2024-04-08 16页
医生护士培训课件:心力衰竭病理诊断治疗
2024-04-08 55页
医疗培训课件:护理护士岗位职责
2024-04-06 35页
医院护士护理培训课件:疼痛管理
2024-04-06 38页
医院医疗保护性约束的基础护理培训课件
2024-04-06 25页
医疗垃圾的分类及处理垃圾分类专题培训
2024-04-06 22页
医院医疗导师带徒工作汇报课件
2024-04-06 17页
医疗培训课件:护士岗位职责
2024-03-21 31页
医疗医院年终工作总结课件
2024-03-21 33页
护理应知应会培训课件
2024-02-20 68页